
https://doi.org/10.1140/epjd/e2003-00151-4
Study of (Al2O3)n(Ox) clusters with n ≤ 16 and x = 0, 1, 2 from first principles calculations
1
Dpto. de Física Teórica, Universidad de Valladolid, 47011
Valladolid, Spain
2
Fachbereich Physik, Universität Osnabrück,
49069 Osnabrück, Germany
3
Dpto. de Física de la Materia
Condensada, C-III, Universidad Autónoma de Madrid, 28049 Madrid, Spain
Corresponding author: a eva@lcb.fam.cie.uva.es
Received:
10
September
2002
Published online:
3
July
2003
The ionic and electronic structure of
(Al2O3)n(Ox) clusters with 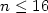 and x = 0, 1, 2
is studied by means of
first principles density functional calculations, norm-conserving
pseudopotentials and a numerical atomic basis set. The equilibrium
geometries have been determined by total energy minimization, starting with
several initial geometries for each cluster size. The trends obtained for
the atomic arrangements (structural isomers, coordination
numbers, “disordered" versus “ordered" structures, etc.) and the
electronic properties (binding energies, Homo-Lumo gap and dipole moments)
are discussed.
For most of the oxidized clusters studied here we find that
the Homo-Lumo gap and the magnitude of dipole moment of isomeric species
can vary drastically.
and x = 0, 1, 2
is studied by means of
first principles density functional calculations, norm-conserving
pseudopotentials and a numerical atomic basis set. The equilibrium
geometries have been determined by total energy minimization, starting with
several initial geometries for each cluster size. The trends obtained for
the atomic arrangements (structural isomers, coordination
numbers, “disordered" versus “ordered" structures, etc.) and the
electronic properties (binding energies, Homo-Lumo gap and dipole moments)
are discussed.
For most of the oxidized clusters studied here we find that
the Homo-Lumo gap and the magnitude of dipole moment of isomeric species
can vary drastically.
PACS: 36.40.-c – Atomic and molecular clusters / 36.40.Cg – Electronic and magnetic properties of clusters / 36.40.Jn – Reactivity of clusters / 61.46.+w – Nanoscale materials: clusters, nanoparticles, nanotubes, and nanocrystals
© EDP Sciences, Società Italiana di Fisica, Springer-Verlag, 2003



