
https://doi.org/10.1140/epjd/e2005-00119-4
Structural and electronic properties of C6 cluster
Department of Applied Physics, National University of
Defense Technology, Changsha, 410073, P.R. China
Corresponding author: a jmyuan@nudt.edu.cn
Received:
6
September
2004
Published online:
13
July
2005
First-principles total-energy calculations of
structural properties of the carbon cluster C6 have been made
using the full-potential augmented plane-waves plus local orbital
(APW+LO) method with the generalized gradient approximation (GGA).
Initiated from a hexagonal configuration, we performed geometry
optimization with damped Newton dynamics. The computed ground
state atomic configuration for C6 belongs to a monocyclic
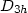 structure. The average bond length is 1.52 a.u. and the
bond angle is
structure. The average bond length is 1.52 a.u. and the
bond angle is 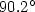 , respectively, which are in agreement with
the reported results.
, respectively, which are in agreement with
the reported results.
PACS: 31.15.Ar – Ab initio calculations / 31.15.Qg – Molecule dynamics and other numerical method / 36.40.-c – Atomic and molecular clusters
© EDP Sciences, Società Italiana di Fisica, Springer-Verlag, 2005


