
https://doi.org/10.1140/epjd/e2005-00034-8
Ab initio study for the intermolecular interaction potential surface of Ar-N2 complex
1
Institute of Atomic and Molecular Physics, Sichuan University, Chengdu 610065, P.R. China
2
International Centre for Materials Physics, Chinese Academy of Sciences, Shenyang 110016, P.R. China
Corresponding author: a xrchen@126.com
Received:
19
October
2004
Revised:
17
November
2004
Published online:
11
April
2005
The intermolecular interaction potentials of van der
Waals Ar-N2 complex have been studied by ab initio calculations using the single
and double excitation coupled cluster [CCSD(T)] theory with perturbative
triples correction. The full counterpoise method is applied to correct the
basis set superposition error (BSSE). It is found that the T-shaped
structure is the most stable conformation with the well depth De of
12.40 meV at the minimum distance Rm of 3.70 Å. The calculated anisotropic
values for  ,
, 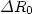 and
and 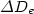 are 0.56 Å,
0.54 Å and 2.68 meV, respectively. Compared with those obtained by others, our
calculated PES seems to be in better agreement with experiments.
are 0.56 Å,
0.54 Å and 2.68 meV, respectively. Compared with those obtained by others, our
calculated PES seems to be in better agreement with experiments.
PACS: 34.20.Gj – Intermolecular and atom-molecule potentials and forces / 31.15.Ar – Ab initio calculations
© EDP Sciences, Società Italiana di Fisica, Springer-Verlag, 2005


