
https://doi.org/10.1140/epjd/e2003-00119-4
On the applicability of deformed jellium model to the description of metal clusters
1
Institut für Theoretische Physik der Universität
Frankfurt am Main, Robert-Mayer 8-10, 60054 Frankfurt am Main, Germany
2
A.F. Ioffe Physical-Technical Institute,
Polytechnicheskaya 26, 194021 St. Petersburg, Russia
Corresponding author: a lyalin@th.physik.uni-frankfurt.de
Received:
10
September
2002
Published online:
3
July
2003
This work is devoted to the elucidation the applicability
of jellium model to the description of alkali cluster properties on the
basis of comparison the jellium model results with those derived
from experiment and within ab initio theoretical framework.
On the basis of the
Hartree-Fock and local-density approximation deformed jellium model
we have calculated
the binding energies per atom,
ionization potentials, deformation parameters and the optimized values of the
Wigner-Seitz radii for neutral and singly
charged sodium clusters with the number of atoms 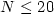 .
These characteristics are compared with the results
derived from the ab initio all-electron simulations
of cluster electronic and ionic structure
based on the density functional theory
as well as on the post Hartree-Fock perturbation theory on many-electron
correlation interaction. The comparison performed demonstrates the great role
of cluster shape deformations in the formation cluster properties and the quite
reasonable level of applicability of the deformed jellium model.
.
These characteristics are compared with the results
derived from the ab initio all-electron simulations
of cluster electronic and ionic structure
based on the density functional theory
as well as on the post Hartree-Fock perturbation theory on many-electron
correlation interaction. The comparison performed demonstrates the great role
of cluster shape deformations in the formation cluster properties and the quite
reasonable level of applicability of the deformed jellium model.
PACS: 36.40.Cg – Electronic and magnetic properties of clusters / 36.40.Mr – Spectroscopy and geometrical structure of clusters
© EDP Sciences, Società Italiana di Fisica, Springer-Verlag, 2003


